LCxLC: sample preparation and measurement
On-line comprehensive RP-LCxRP-LC/IT-TOF for the analysis of proteome isoforms: a meaningful evaluation tool for native and recombinant proteins
Introduction

The analysis of complex biochemical systems, such as proteins and peptides isolated from tissues, cells, and body fluids has always represented a major task for analytical chemistry. A high demand is in fact placed both on the power of separation techniques, given the extremely high complexity of the proteome samples, and on the sensitivity of detection methods, to enable probing of low abundant proteins or peptides. Different strategies have been developed attempting to address the needs of modern proteomics, increase the overall throughput of proteomics experiments, and facilitate researchers to investigate into the complicated biological networks in which proteins are involved, at different levels.
For many years, two-dimensional polyacrylamide gel electrophoresis (2D-PAGE) followed by mass spectrometry (MS) has been the workhorse in analytical proteomics, its major drawbacks consisting in difficulty of automation, low accessibility of membrane-bound proteins, problematic detection of proteins with large molecular weight, high pI, strong hydrophobicity, or low abundance. Over the last decade, considerable effort has been put in the development of ultra-high efficiency liquid chromatographic (LC) methodologies, pushing gel-free separation techniques to evolve beside the more troublesome and tedious 2D-PAGE experimental designs.
2D separation set-ups based on RP-LC are characterized by superior resolution and peak capacity, more homogeneous distribution of peptides elution in the separation window, robustness and easy handling.
This technical report describes the first comprehensive 2D LC system, in which the on-line coupling of RP-LCxRP-LC to MS (IT-TOF) is investigated. The two dimensions consisted both of a novel fused-core stationary phase specifically designed for peptide separation, interfaced through an electronically activated 2 position, ten-port valve. The performance of the system was assessed by means of tryptic mapping (protein unfolding, trypsin digestion, and reversed-phase chromatography of the peptide samples), followed by ESI-MS characterization of α-casein and dephosphorylated α-casein.
LCxLC-IT-TOF MS analyses
Sample preparation
For the preparation of the tryptic digest, the procedure was based on previous work of Bushey and Jorgeson, one-tenth gram of α-casein or dephosphorylated α-casein were dissolved in 10 mL of 0.01 M HCOONH4 buffer, and the pH adjusted to 8.0 with NH4OH; the solution was heated in a boiling water bath for 6 min.
After the solution cooled, 2.0 mg of trypsin from bovine pancreas were added, and the mixture was allowed to react for 4 h at + 37 °C; the reaction was quenched by adding 0.1% TFA to pH 2. The digests were stored at + 4 °C, and filtered prior to injection through 0.45 μm nylon membrane (Whatman). Aqueous solutions of the peptide standard mixture (500 ppm) were injected without any filtration.
Instrumentation
Comprehensive 2D-LC analyses were performed on a Shimadzu Prominence LC-20A (Shimadzu, Milan, Italy), consisting of a CBM-20A controller, two LC-20AD dual-plunger parallel-flow pumps (D1-LC), an LC-20 AB solvent delivery module, equipped with two dual- plunger tandem-flow pumps (D2-LC), a DGU-20 A5 degasser, an SPD-M20A photo diode array detector (8 μL detector flow cell volume), a CTO-20A column oven, a SIL-20A autosampler. The two dimensions were connected by using an electronically controlled 2-position ten-port high pressure switching valve (Rheodyne, supplied by Shimadzu, Kyoto, Japan), placed inside the column oven and equipped with two empty 0.254 mm I.D. PEEK loops of identical volume (100 μ0L). Both dimensions and the switching valve were controlled by the LCMSsolution software (Version 3.50.346, Shimadzu). The LCxLC data were visualized and elaborated into two- and three dimensions using Chromsquare ver. 2.0 software (Chromaleont, Messina, Italy). The LCxLC system was coupled to an IT-TOF mass spectrometer through an ESI source (Shimadzu, Kyoto, Japan).
Analytical conditions
First dimension (1D) column: Ascentis Express Peptide ES-C18, 150 mmL. x 2.1 mm I.D., 2.7 μm d.p. (Sigma-Aldrich/Supelco, Bellefonte, PA, USA)
Mobile phase: (A) 10mM CH3COONH4 in H2O (pH 9); (B) 10mM CH3COONH4 in H2O/ACN 10:90 (pH 9)
Gradient: 0-40 min, 0 to 10% B, 40-60 min, to 20% B, 60-200 min, to 50%, 200-220 min, to 100% B (hold for 20 min)
Flow rate : 100 μL/min
Column oven : 35 °C
Injection vol.: 20 μL
Second dimension (1D) column: Ascentis Express Peptide ES-C18, 30 mmL. x 4.6 mmI.D., 2.7 μm d.p. (Sigma-Aldrich/Supelco, Bellefonte, PA, USA)
Mobile phase: (A) 0.1% TFA in H2O (pH 2); (B) 0.1% TFA in H2O/ACN 10:90 (pH 2)
Gradient : 0-0.05 min, 0 to 20% B, 0.05-0.40 min, to 40% B, 0.40-0.50 min, to 50% B, 0.50-0.69 min, to 90% B, 0.69-0.70 min, to 0% B, 0.70-1.00 min, to 0% B
Flow rate : 4 mL/min
Column oven : 35 °C
Modulation time : 1 min
Results and discussion
The contour plot obtained for the comprehensive RP-LCxRP-LC/PDA/IT-TOF analysis of α-casein and dephosphorylated α-casein is shown in Figure 1A-B.
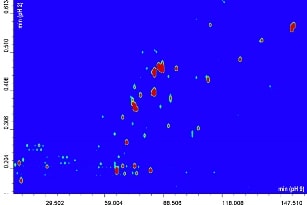
Figure 1A
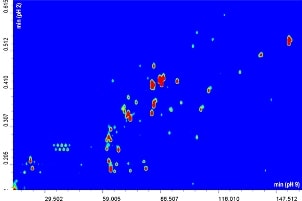
Figure 1B
Figure 1A-B. 2D RPxRP-LC plot of α-casein (A) and dephosphorylated α-casein (B) tryptic digest separation. Colours are used to represent the value of absorbance at a given point. The relation between colours and absorbance is ruled by a colour map, that can be configured by the user.
Replicate analyses (n=3) of the digests were run on the 2D system, and reproducibility of the retention times was calculated for five selected peaks spread throughout the elution window, yielding an average CV% of 0.555 (averaged CV% in the two chromatographic dimensions). The overall peak capacity of the comprehensive separation was calculated as 8540, being multiplicative of the individual values obtained for the two dimensions (n1xn2). Figure 2 shows a five-minute enlargement of the raw RP-LCxRP-LC/PDA/IT-TOF chromatogram corresponding to the plot in Figure 1A-B, and the average mass spectrum obtained for three consecutive peaks (1 min interval, corresponding to the modulation time set) are depicted in the inset.
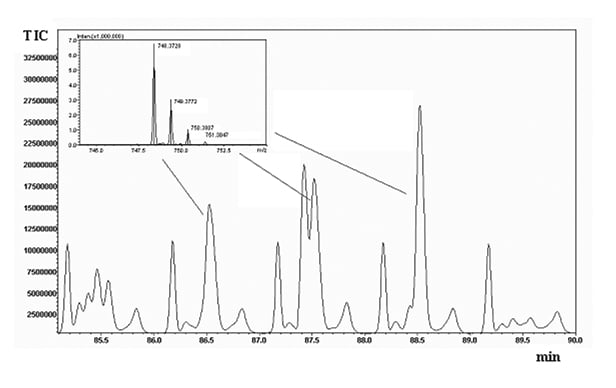
Figure 2. Five-minute enlargement of the raw chromatogram obtained for the 2D RPxRP-LC separation of -α casein tryptic digest (plot in Figure 1A). The inset shows m/z values corresponding to three consecutive cuts from the first dimension.
This demonstrates that at least three fractions of the peptide with m/z 747.3650 have been transferred from the first to the second dimension of the comprehensive system. The acquired mass spectrometry data were manually processed, by averaging the number of scans within each chromatographic peak, and deconvolution of charge envelope was afterwards performed for [M+H]+ and [M+nH]n+ ions. The experimental deconvoluted molecular masses of the peptides were then compared to the theoretical values obtained by in-silico digestion of the proteins, to obtain the relative sequence coverage.
In-silico digestion of α-casein and dephosphorylated α-casein was performed by using PeptideMass software available at Expasy site (www.expasy.ch/tools/peptide-mass.html), selecting up to three missed cleavages (MC) for the generation of peptides. For each identified peptide, monoisotopic molecular masses of the phosphorylated and the corresponding dephosphorylated forms are reported, together with the position of the modified aminoacidic residues.
Many of the differences in the plots in Figure 1A-B are obviously related to the presence or absence of phosphorylated peptides.
Since phosphorylation occurs on the serine residue, and a difference of (roughly) 80 Da was observed for removal of each phosphate group, it must be conclusive that only HPO3 - is removed by dephosphorylation, leaving the serine residue intact. By combining the results obtained from the two RP LC dimensions, sequence coverage of 90.3% and 76.3% for α-casein and dephosphorylated α-casein, respectively, were obtained. Comparison with the corresponding values of 68.2% and 56.4% obtained for the mono-dimensional system (1D, four coupled columns) clearly demonstrates the usefulness of the 2D-LC system.